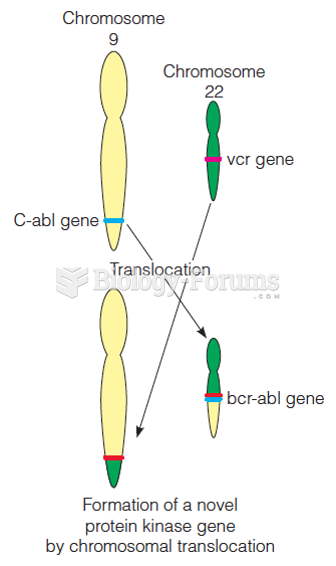
In another word, I want to know the protein domain(s) that contains that nsSNP.
Hello
ahmedmelmoselhy, here are some articles I found that'll help you here...
Non-synonymous Single Nucleotide Polymorphisms (nsSNPs) potentially disrupt normal protein functions. Several computational tools exist today that can predict the damaging effects of nsSNPs on protein structure and functions. For example, PolyPhen predicts the result of amino acid substitutions on the structure and function of human proteins (Adzhubei et al., 2010). The impact of these tools is enormous on clinical interpretation of human nsSNPs but no such tool is available for the model organism yeast although SNPs are largely available and being identified in large-scale experiments. The most helpful tool to interpret nsSNPs in yeast is the SGD Variant Viewer (Cherry, 2015), but it does not predict impact on structure and function. Hence a tool to analyze the effect of nsSNPs on different functional regions of yeast proteins is much needed to accelerate research in yeast systems biology. We chose to develop such a tool and particularly focus on Post-Translational Modications (PTMs) and protein domains affected by nsSNPs. UniProt (The UniProt Consortium, 2014) contains over 200 different types of PTMs. PTMs can modulate biological interactions and broadly impact on protein functions, stability and localization hence phenotypes in response to stimuli. An nsSNP at a PTM residue under normal or experimental conditions could explain a change in phenotype and could thus be important for interpretation of effects of nsSNPs (Reimand et al., 2015).
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5408805/The QTL region associated with AHR was also mined for candidate genes that contain sequences variants that were predicted to have functional effect at the protein level. Within our QTL we identified 50 coding nsSNPs in 27 different genes that are possible candidates for causing variation in the AHR trait (Table 4). To reduce the number of candidates we used amino acid substitution prediction tools, PROVEAN and PolyPhen2, to determine which coding nsSNPs may affect the protein function from those that are neutral. Each tool uses its own algorithm based on protein sequence and/or structure to determine the effect. PROVEAN compares the mutated sequence to the reference sequence to calculate a delta alignment score. The score predicts the effect of an amino acid substitution in the context of its flanking sequences. A score of −2.5 was determined by PROVEAN creators as a default cutoff. Substitutions with scores less than −2.5 are considered deleterious, while substitutions with scores less than −4.1 are of greater confidence [19]. PolyPhen2 describes allele function as “benign”, “possibly damaging”, or the most confident, “probably damaging”. If a prediction cannot be made due to lack of sequence alignment data, then it is reported as “unknown”. Polyphen2 is based on comparison of sequence homology, three dimensional structure, and SWISS-PROT annotation of protein domains [20]. Genes with nsSNPs whose predicted effects are in consensus between both tools were selected as candidates.
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0104234Non-synonymous single nucleotide polymorphisms (nsSNPs) can potentially alter the structure and the function of the proteins, and thus are excellent candidates for complex disease association studies. We had previously extracted a total of 85 nsSNPs from 42 cell cycle genes from 5 public SNP databases following a highly stringent SNP extraction procedure. All these nsSNPs were analyzed by the SIFT tool (Ng and Henikoff, 2002) for their evolutionary conservation status. Here, we present categorization of the nsSNPs based on their locations on functional protein domains and motifs. For this purpose, we utilized the SWISS-PROT (O’Donovan et al. 2002) and the InterPro (Mulder et al. 2003) protein databases to identify the nsSNPs located in protein domains. Short linear protein motifs involved in protein-protein recognition/interactions processes were predicted using the ScanSite 2.0 tool (Obenauer et al. 2003). As a result, a total of 37 (43.5%) nsSNPs were found located in a protein domain. Thirteen of these nsSNPs were evolutionary conserved, suggesting that these nsSNPs may directly affect the protein function. The minor allele frequency of one of these nsSNPs (CDK6-R31S) was reported as 29% in one of the SNP databases. Another variation at that position (R31C) of CDK6 was already reported to cause disruption of its binding to INK proteins and localize the CDK6 protein in the cytoplasm (Grossel et al. 1999), which makes CDK6-R31S an excellent candidate for association studies in cancer. A total of 8 (9.4%) nsSNPs were located in a short protein motif. Among them, 3 nsSNPs caused abolishment of a SH2 (in Cyclin B1) and two protein kinase recognition motifs (in Cyclin G1 and CDKN1A). The nsSNP in Cyclin B1 occurred at an evolutionary conserved residue, suggesting that it may interfere with the proper recognition/interaction of this protein. Verification of these predictions by means of elaborate experimental approaches is underway. To sum up, here we present the results of a bioinformatics-based analysis to select the SNPs with a probable impact on the protein function. Those nsSNPs that are both evolutionary conserved and located in a function protein domains/motifs are excellent candidates for cancer-association as well as functional studies.
https://cancerres.aacrjournals.org/content/64/7_Supplement/1109.1 [not free]SIFT predicts whether an amino acid substitution affects the protein function based on sequence homology and the physical properties of amino acid.
https://shodhganga.inflibnet.ac.in/bitstream/10603/37798/13/13_chapter%204.pdfIt appears that most of these articles are mentioning
SIFT method (Sorting Intolerant from Tolerant).

 Quick Reply
Quick Reply